
Summary
This blog explains why manual batch record review is becoming a serious problem in pharmaceutical manufacturing. Most pharma companies still rely on paper records or static PDFs, which forces QA teams to spend large amounts of time checking signatures, dates, calculations, and documentation manually.
The article describes this as the “Hidden Plant” because companies invest huge amounts of effort into administrative review work that does not directly improve product quality or patient outcomes. These manual processes slow down batch release, increase operational costs, delay inventory movement, and create compliance risks.
The blog also explains how regulators are now encouraging a shift toward Review by Exception (RbE). In this approach, software automatically reviews compliant data and only sends unusual events or deviations to QA teams for investigation. This helps quality teams focus on real risks instead of repetitive verification work.
Finally, the article highlights that different pharma organizations need different approaches to modernization. Virtual pharma companies, CDMOs, and large manufacturers all operate differently, so the right digital strategy depends on the organization’s structure, systems, and level of digital maturity.
The Hidden Plant: An Invisible Burden
Right now, somewhere in your organization, a QA reviewer is working through page 87 of a 180-page batch record. She is verifying, for the third time, that a date format is correct, a signature is present, and a yield calculation matches the preceding step. Tomorrow, she will do it again. The day after that, the same.
Pharmaceutical manufacturing has a name for this invisible burden. Practitioners call it the Hidden Plant, also known as the Paper Plant. The Hidden Plant is the massive, unseen administrative infrastructure of documentation, routing, signing, and reviewing required to keep a manufacturing facility legally compliant. It absorbs as much organizational labor as the physical transformation of raw materials into medicine. And it generates zero patient value.
A standard Master Batch Record for a complex therapeutic can span 150 to 200 pages, containing thousands of individual data entries, material lots, calculations, and operator signatures. The regulatory ground beneath the systems that govern this process is shifting. The question is no longer whether to modernize batch review. The question is which digital pathway fits your organization’s actual operating model. That answer is not uniform across organizational archetypes.
The Numbers Behind the Burden
Industry estimates suggest that 60 to 70 percent of pharmaceutical manufacturers and CDMOs remain heavily reliant on manual, paper-based review processes or static digital equivalents. That figure is more than a data point about technology adoption. It reflects a structural inertia built over decades, compounded by misplaced investment and a persistent misreading of what digitization actually means.
Operational consequences are measurable. In complex biologics manufacturing, batch record review alone can take up to 15 days from production completion to final quality approval. When deviations or out-of-specification events occur, investigations can add another 5-15 days to the release cycle. These delays directly impact inventory availability, working capital, and supply responsiveness, especially in high-volume or high-value manufacturing environments.
Industry data suggests that approximately 50 percent of batch record problems originate not from manufacturing deviations, but from human documentation errors. Read that again: the QA review process is catching its own administrative mistakes at roughly the same rate it is catching process failures. Manual review is not revealing production problems. It is consuming expert capacity to find transcription errors.
Misallocation runs deeper. In paper-reliant facilities, right-first-time rates have been reported as low as 47 percent. Quality assurance professionals are spending upwards of 70 percent of their operational bandwidth verifying non-critical data (date formats, blank fields, calculation carry-overs) rather than investigating root causes or driving process improvements.
Many organizations believe they have solved this problem by scanning paper records into PDF format and storing them electronically. They have not. Paper-based data capture is a digital dead end. It is the practice of digitizing documents for storage while leaving the underlying manufacturing data unstructured and unqueryable. A system that holds a PDF cannot execute an algorithmic exception rule. It cannot detect a missing signature. It cannot trend a parameter across batches. Storing documents is not the same as governing data.
Operational burden alone would justify action. Three regulatory forces are now making the status quo a compliance liability as well.
Three Regulatory Forces Are Closing the Window
Review by Exception (RbE), also known as Exception-Based Review, Automated Batch Disposition, or Exception Reporting, is a risk-based data review methodology recognized by ISPE and the Parenteral Drug Association (PDA). It defines a process in which quality personnel examine only information, audit trail entries, and process events that deviate from predefined, validated parameters. Rather than performing exhaustive line-by-line verification of compliant data, RbE systems algorithmically evaluate batch records against specifications and control limits, flagging anomalies for human intervention and passing clean data through automatically.

Three converging regulatory forces are transforming RbE from an operational best practice into a compliance expectation. The July 2025 draft revision of EU GMP Annex 11, released by the European Medicines Agency, formalizes Review by Exception within regulatory text for the first time. As of Q2 2026, the draft remains in consultation, and final text may differ. The directional intent is unambiguous: computerized systems must be technically capable of detecting boundary excursions before RbE can be legally utilized.
The FDA’s Quality Management Maturity (QMM) program, now in its third year of voluntary site assessments, has publicly stated that the transition from manual data verification toward algorithmic oversight is a key indicator of organizational maturity. Organizations replacing mechanical proofreading with structured exception review position themselves favorably for QMM assessment; those relying on paper accumulate a maturity gap that will be increasingly difficult to close.
Enforcement data underscores the urgency. Fiscal year 2025 brought 112 CGMP warning letters citing deficiencies under 21 CFR 211, the highest volume in over two decades Editorial analysis estimates that approximately 21 percent of all FDA warning letters stem from poor documentation practices. These are not violations caused by bad manufacturing. They are violations caused by inadequate data governance.
RbE applies Quality by Design thinking to the documentation layer. It engineers the system so compliant records flow through automatically, and human expertise concentrates on the exceptions that genuinely threaten product quality and patient safety. That is the transition Quality by Inspection has never been able to deliver.
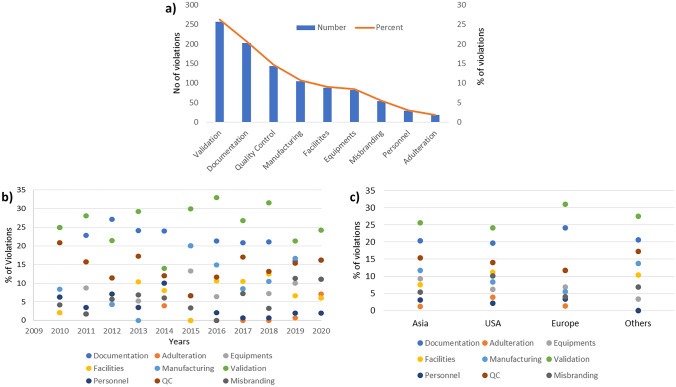
a) Sub-categorization of violations observed in CGMP warning letters issued to pharmaceutical manufacturers and its trend analysis based on b) year and c) regions
Source:https://pmc.ncbi.nlm.nih.gov/articles/PMC9377664/
The Math of the Paper Plant
Direct cost of manual batch review is calculable. Take your organization’s annual batch volume. Multiply by the industry benchmark of approximately 48 active working hours of QA labor per complex therapeutic batch. Apply your fully loaded labor cost per hour. The resulting figure captures only the direct review burden. It is consistently sobering when organizations run it against their own numbers.
Most internal business cases stop there. They are wrong to. Direct review burden captures only the labor your QA team logs against batch review itself. It misses the working capital trapped in quarantine, the rework loop that consumes your most experienced reviewers, and the strategic work your QA leaders cannot perform because their calendars are full of mechanical verification.
Add the inventory carrying cost of finished goods sitting in quarantine for 10 to 40 days awaiting release. Add the rework costs driven by a right-first-time rate as low as 47 percent in paper-reliant environments. Add the rework costs driven by a right-first-time rate. Every batch that does not pass first-time review requires remediation, investigation, and re-review. Add the opportunity cost of highly compensated QA professionals whose bandwidth is consumed by mechanical data verification rather than root-cause investigation, process improvement, or regulatory strategy.

The whitepaper, A Quality Leaders Guide to Review by Exception provides the complete methodology, including carrying cost estimation, rework cost quantification, and the framework for building a defensible internal business case with your own operational data.

What Comes Next
Operational burden. Regulatory pressure. Financial exposure. The convergence of all three makes the status quo increasingly untenable for pharmaceutical organizations of every size and operating model. The question is not whether to act. It is how. And the answer is not uniform.
Different organizations face structurally different barriers to modernizing batch review. A virtual pharmaceutical sponsor managing CDMO relationships operates under entirely different constraints than an integrated manufacturer bridging fragmented legacy systems. A virtual sponsor managing CDMO records faces a different decision than a CDMO scaling multi-client production. A mid-market manufacturer moving beyond paper-based capture faces a different decision than a fully integrated producer with capital for full-stack deployment. Digital data governance architectures that address those constraints are not one-size-fits-all. The right starting point depends on where your organization stands today and what it can realistically deploy. The pathway that fits is the one matched to your reality.
In a next post, we examine three digital architectures for achieving Review by Exception, each designed for a different organizational reality, from paper-dependent virtual pharma sponsors to fully integrated manufacturers.






